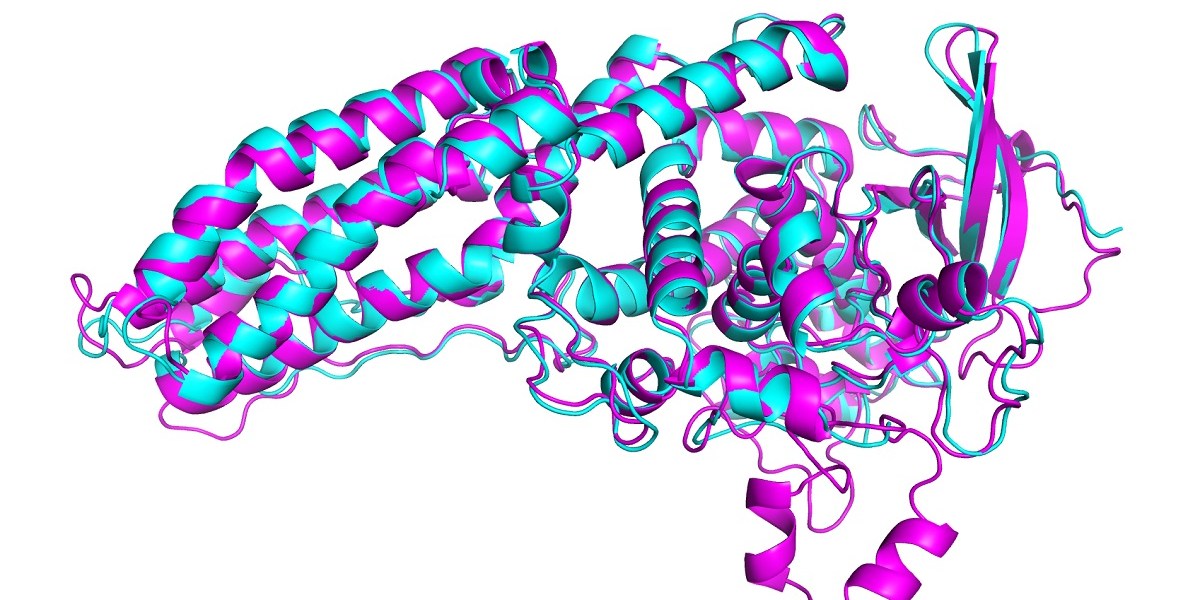
En el CASP de este año, AlphaFold predijo la estructura de docenas de proteínas con un margen de error de solo 1,6 angstroms, es decir, 0,16 nanómetros. Esto supera con creces todos los demás métodos computacionales y, por primera vez, coincide con la precisión de las técnicas experimentales para trazar la estructura de las proteínas en el laboratorio, como microscopía crioelectrónica, resonancia magnética nuclear y Cristalografía de rayos X. Estas técnicas son costosas y lentas: pueden costar cientos de miles de dólares y años de prueba y error para cada proteína. AlphaFold puede encontrar la forma de una proteína en unos pocos días.
Pero identificar la estructura de una proteína es muy difícil. Para la mayoría de las proteínas, los investigadores tienen la secuencia de aminoácidos en la cinta, pero no la forma retorcida en la que se pliegan. Y normalmente hay una cantidad astronómica de formas posibles para cada secuencia. Los investigadores han estado luchando con el problema al menos desde la década de 1970, cuando Christian Anfinsen ganó el premio Nobel por demostrar que las secuencias determinaban la estructura.
El avance podría ayudar a los investigadores a diseñar nuevos medicamentos y comprender las enfermedades. A largo plazo, predecir la estructura de las proteínas también ayudará a diseñar proteínas sintéticas, como las enzimas que digieren los desechos o producen biocombustibles. Los investigadores también están explorando formas de introducir proteínas sintéticas que aumentarán el rendimiento de los cultivos y harán que las plantas sean más nutritivas.
Impactante avance
“Es un avance muy sustancial”, dice Mohammed AlQuraishi, biólogo de sistemas de la Universidad de Columbia que ha desarrollado su propio sistema para predecir la estructura de las proteínas. “Es algo que simplemente no esperaba que sucediera tan rápido. Es impactante, en cierto modo “.
“Esto realmente es un gran problema”, dice David Baker, director del Instituto de Diseño de Proteínas de la Universidad de Washington y líder del equipo detrás de Rosetta, una familia de herramientas de análisis de proteínas. “Es un logro asombroso, como lo que hicieron con Go”.
El lanzamiento de CASP en 1994 dio un impulso al campo de la predicción de estructuras de proteínas. Cada dos años, los organizadores publican aproximadamente 100 secuencias de aminoácidos para proteínas cuyas formas se han identificado en el laboratorio pero aún no se han hecho públicas. Luego, decenas de equipos de todo el mundo compiten para encontrar la forma correcta de plegarlos utilizando software. Muchas de las herramientas desarrolladas para CASP ya son utilizadas por investigadores médicos. Pero el progreso fue lento, con dos décadas de avances incrementales que no lograron producir un atajo al trabajo de laboratorio minucioso.
Cuando DeepMind ingresó a la competencia en 2018 con su primera versión de AlphaFold, le dio a CASP la sacudida que estaba buscando. Todavía no podía igualar la precisión de un laboratorio, pero dejó otras técnicas computacionales en el polvo. Los investigadores tomaron nota: pronto muchos estaban adaptando sus propios sistemas para trabajar más como AlphaFold.
Este año, más de la mitad de las entradas utilizan alguna forma de aprendizaje profundo, dice Moult. Como resultado, la precisión general fue mayor. El nuevo sistema de Baker, llamado trRosetta, usa algunas de las ideas de DeepMind de 2018. Pero aún llegó en un “segundo lugar muy distante”, dice.
En CASP, los resultados se puntúan utilizando lo que se conoce como prueba de distancia global (GDT), que mide en una escala de 0 a 100 qué tan cerca está una estructura predicha de la forma real de una proteína identificada en experimentos de laboratorio. La última versión de AlphaFold obtuvo una buena puntuación para todas las proteínas del desafío. Pero obtuvo una puntuación GDT superior a 90 para alrededor de dos tercios de ellos. Su GDT para las proteínas más duras fue 25 puntos más alto que el del siguiente mejor equipo, dice John Jumper, quien dirige el equipo AlphaFold en DeepMind. En 2018, la ventaja rondaba los seis puntos.